Drug Repurposing for Alzheimer's Disease Using Graph Neural Networks
All analyses done using Python.
Introduction
Historically, neural networks in drug discovery used the fixed chemical properties of molecules as feature input into neural networks. These networks could then predict the efficacy/activity of a suggested compound given its chemical properties.
In 2020, Collins and colleagues [1] were able to utilize graph convolutions to produce trainable molecular features for use in their neural networks. These trainable features were then fed into a feed-forward neural network to predict the antibiotic activity of a compound against E. coli. This new utilization of graph neural networks to train molecular features led to the discovery of a novel antibiotic, one that was structurally unique to other antibiotics.
The use of neural networks to predict novel treatments of disease is widespread and includes many areas of medicine and disease. Alzheimer's Disease (AD) is one of the most impactful and prevalent diseases in the world, impacting an estimated 55 million people worldwide. Many hypotheses involving different genes, proteins, and other factors have been proposed as contributing to AD, however, two proteins have become notoriously associated with AD: the Tau protein and A-beta amyloid peptide (APP) [2, 3]. These two proteins contribute to the neurofibrillary tangles and amyloid plaques present in the brains of AD patients. Although considerable research has shown relationships between Alzheimer’s Disease and the dysfunction of these two proteins, there is little evidence that we can successfully reverse the dysregulation of these two proteins in-humans, with hopes of preventing the onset or the progression of the disease.
Using drugs pulled from the DrugBank Database [4], this project aims to employ the same strategy Collins and colleagues used to identify novel antibiotics, but with the goal of identifying drugs for the repurposed use of treating Alzheimer’s Disease.
Representing Molecules through Bits and Graphs
Many different identifiers have been developed for compounds, including the InChi, InChiKey, and SMILES. These identifiers are useful in different contexts, however, are not useful for most machine learning models. The Morgan Fingerprint was developed as a hashed representation of compounds that serve as fixed-length bit vectors (1s and 0s) in which molecular structures can be deduced. Typically, Morgan Fingerprints are generated in lengths of 1,024 or 2,048, with longer vectors providing more detailed structural breakdown. These bit vectors can be passed into machine learning models before being passed through and transformed within the model’s hidden layers. When generating a Morgan fingerprint, the substructures encoded within a vector are dependent on the fixed “radius” parameter encoding how many adjacent atoms are considered for substructure generation. This parameter limits the structural “awareness” across the molecule regarding the presence of functional groups that may be important when present further apart in the molecule. The image below shows fingerprints encoded with a radius of 2, where the encoded structures are built from neighboring atoms at most 2 steps away from the from the central atom highlighted in blue.
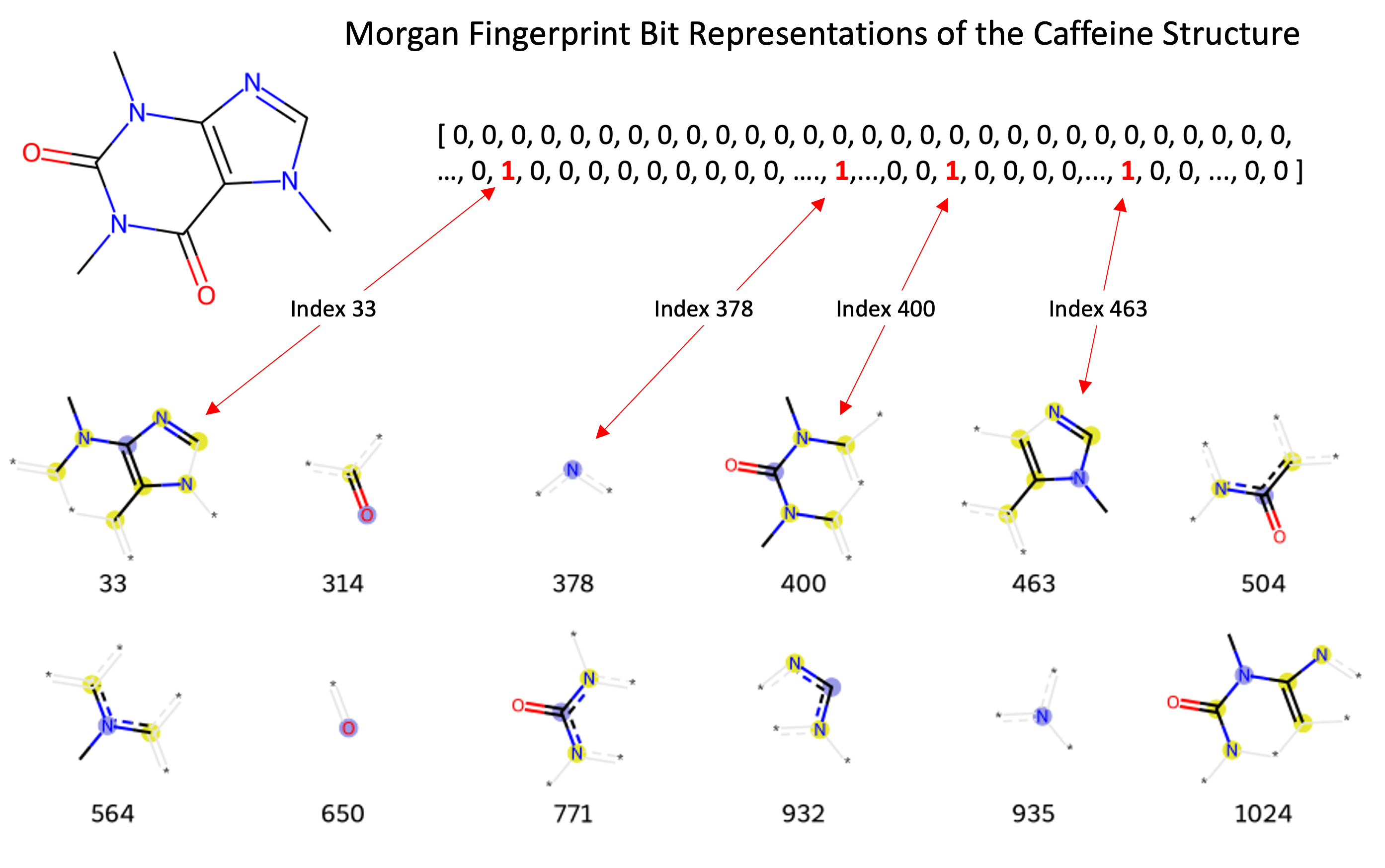
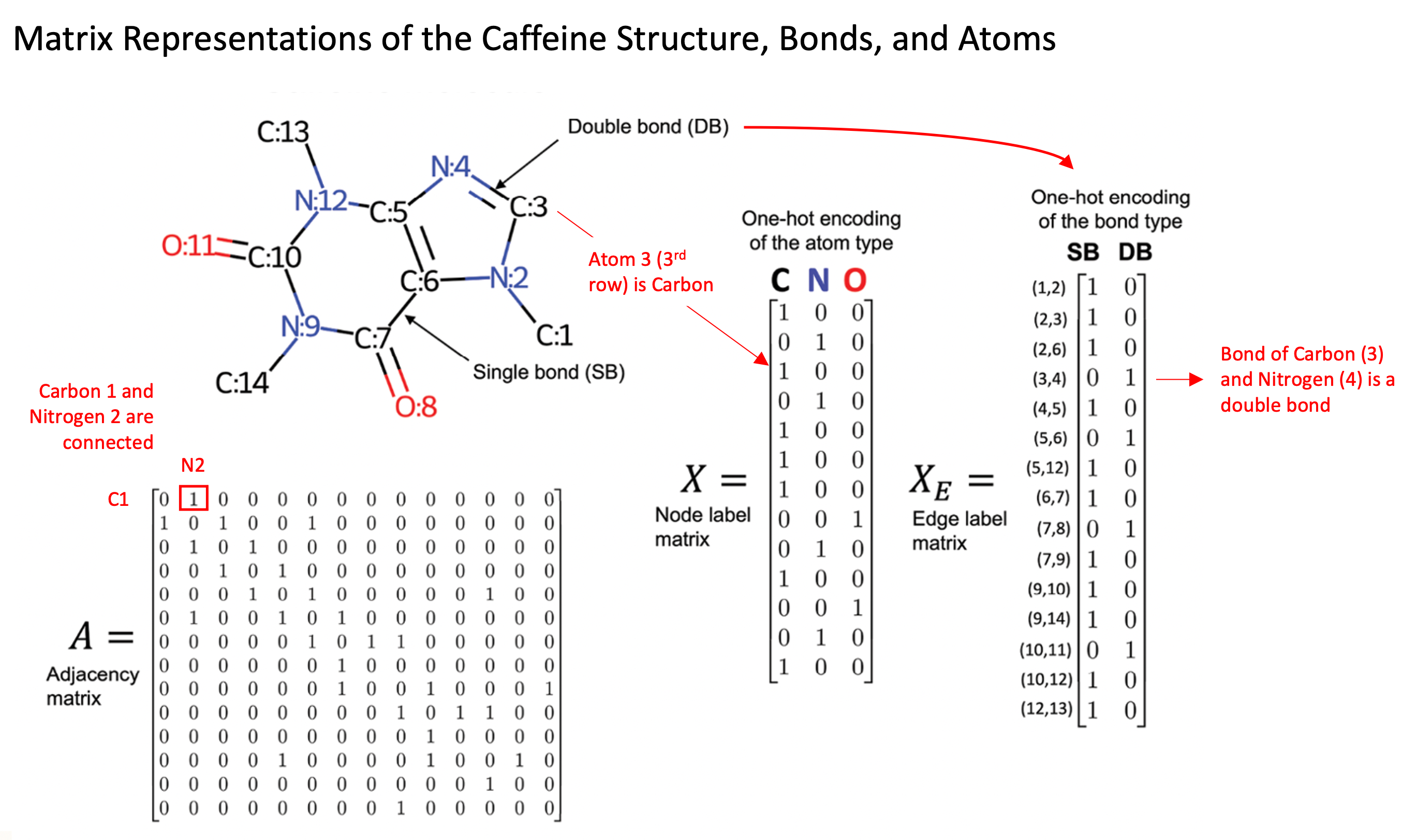
Although fingerprint representations of molecules are useful in some contexts, a more recent development is the representation of molecules as graph structures, where atoms are presented as nodes and bonds between atoms as edges. The use of these graph representations provides several advantages over fixed Morgan Fingerprints, such as the ability to learn spatial arrangements of atoms and to encode atomic and molecular features explicitly. The image to the [direction] illustrates the caffeine molecule represented through the adjacency matrix, node (atom information) matrix, and edge label (bond information) matrix. These matrices allow us to explicitly represent specific features of each individual atom and bond, such as the type of atom, number of protons, number of neighboring atoms, and bond types.
Graph Convolutional Neural Networks
Graph Convolutional Neural Networks (GCN) are a specific type of Graph Neural Network, and build on many of the features of Convolution Neural Networks (CNN) typically used on images. Graphs however serve as more flexible structures that more easily handle variations in input data, in comparison to CNNs which largely work with fixed size image vectors (i.e. 64 x 64-pixel images). The ability to handle variable size inputs is essential for GCNs to fully evaluate molecules. Current small molecule drugs are as small as a single atom (i.e. Lithium) and grow upwards of 37 heavy atoms [5], providing an enormous-range of potential graph sizes and shapes.
Many different GCN architectures exist, but are often in general composed of input layers, hidden graph convolutional layers, pooling layers, and subsequent feed forward / fully connected layers to provide predicted outputs.
The graph convolutional layers are one feature that makes GCN’s unique and especially useful. These layers lead to the model learning of expanded neighborhoods of atoms, known as message passing. As nodes are often only connected to a small subset of other nodes in the graph set, a single graph convolution layer only incorporates information from nodes a single step away. This is when multiple graph convolution layers become useful, as each additional layer allows the passing of information from one node “further”, enabling more distance nodes to influence the values of a node of interest deeper in the network.
For a more explicit example, we can consider this in respect to the caffeine molecule in the image above. Each atom’s encoding will be updated at each layer of the network. When Carbon C:7 is updated in the first graph convolution layer, it will aggregate information from its adjacent neighbors, Oxygen O:8, Nitrogen N:9 and Carbon C:6. When the Nitrogen N:9 is updated, it will take aggregate data from Carbon C:10 and Carbon C:14. This will be done for each atom in the molecule to complete the first convolutional layer. In the second convolutional layer, when Carbon C:7 is aggregating information from its neighbors, information from the updated Nitrogen N:9 will represent the original Nitrogen atom, but with influence from its own adjacent neighbors, Carbons C:10 and C:14. Therefore, this convolutional layer led Carbon C:7 to learn from atoms another step further away than its immediate neighbors. As the number of convolutional layers increases, each layer increases the number atoms away that the updated value accounts for.
Another important feature of graph convolutional networks are the pooling layers. The pooling layers can be of different types, including mean, min, and max pooling. These all serve the purpose to consolidate information from a neighborhood of atoms, with each type emphasizing different values within a neighborhood. In the case of max pooling, the pooling layer will extract the maximum feature values out of adjacent atoms in each neighborhood, emphasizing the most prominent atomic features. These features may include properties such as the number of bonds connected to each atom, the number of protons, the chirality of the molecule, or the atom type.
The last layer or layers of a graph neural network is typically a feed-forward (dense) layer, in which the logits of the model are produced and fed into a final softmax function. In a classification task, this softmax function produces the final class probabilities, which are then classified based on the chosen classification threshold. In this project, the class probabilities are split between two binary labels: whether the molecule will inhibit the expression of Tau or APP.
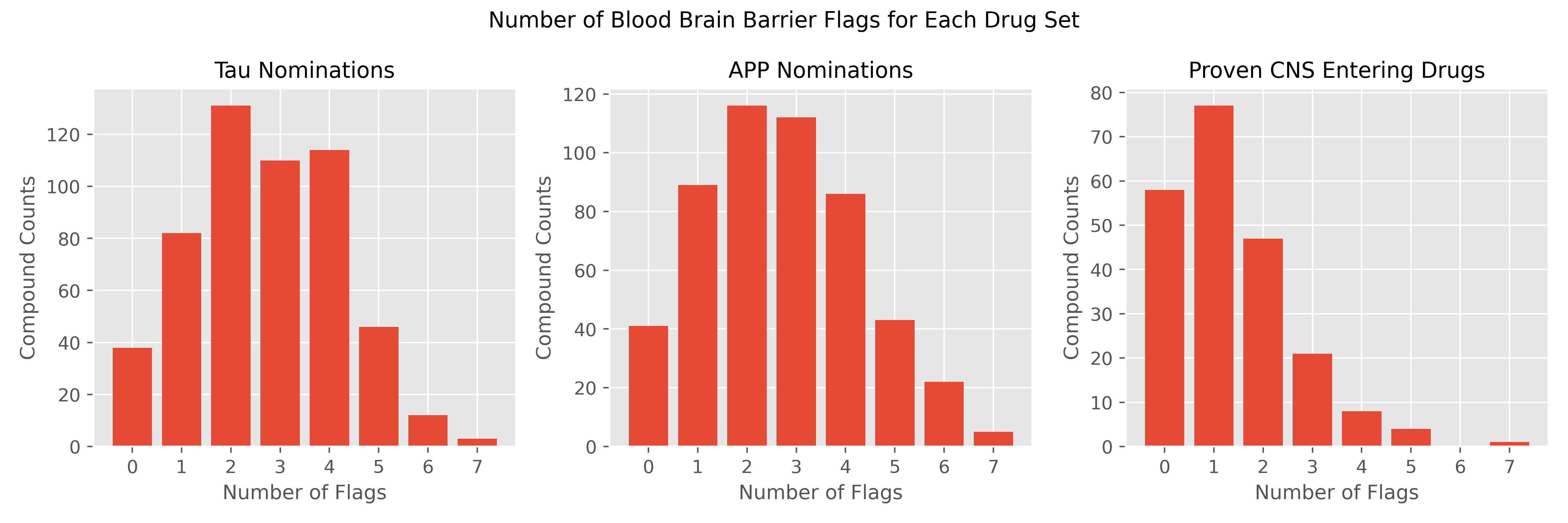
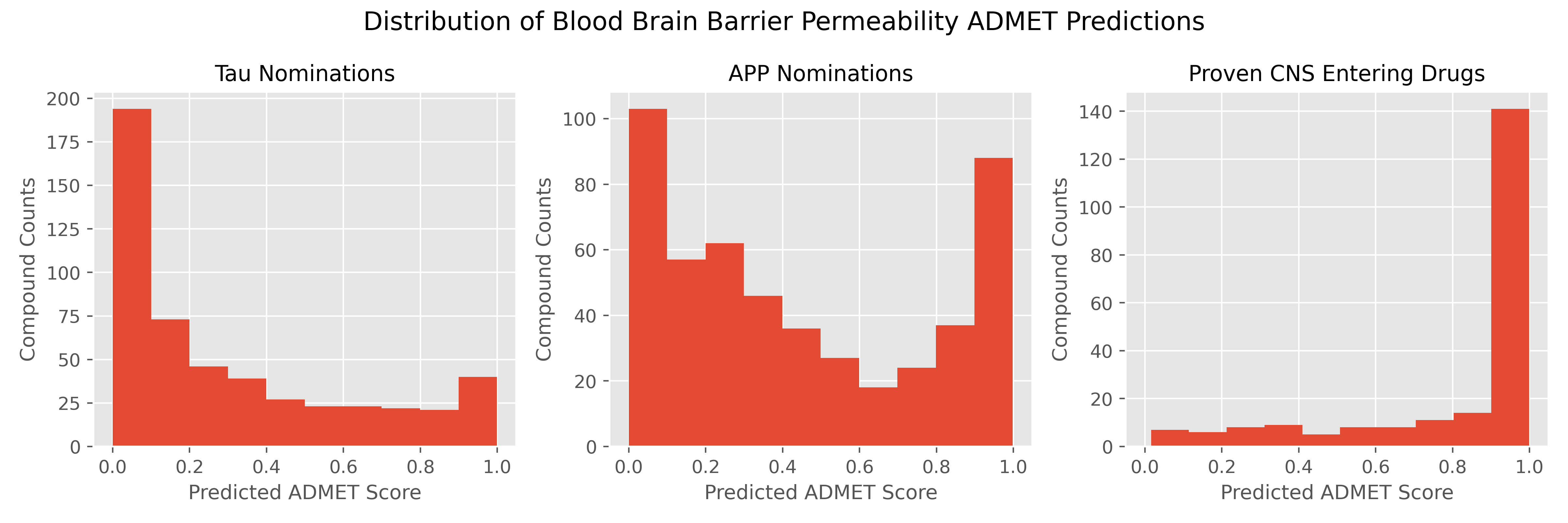
An Introduction to the Blood Brain Barrier
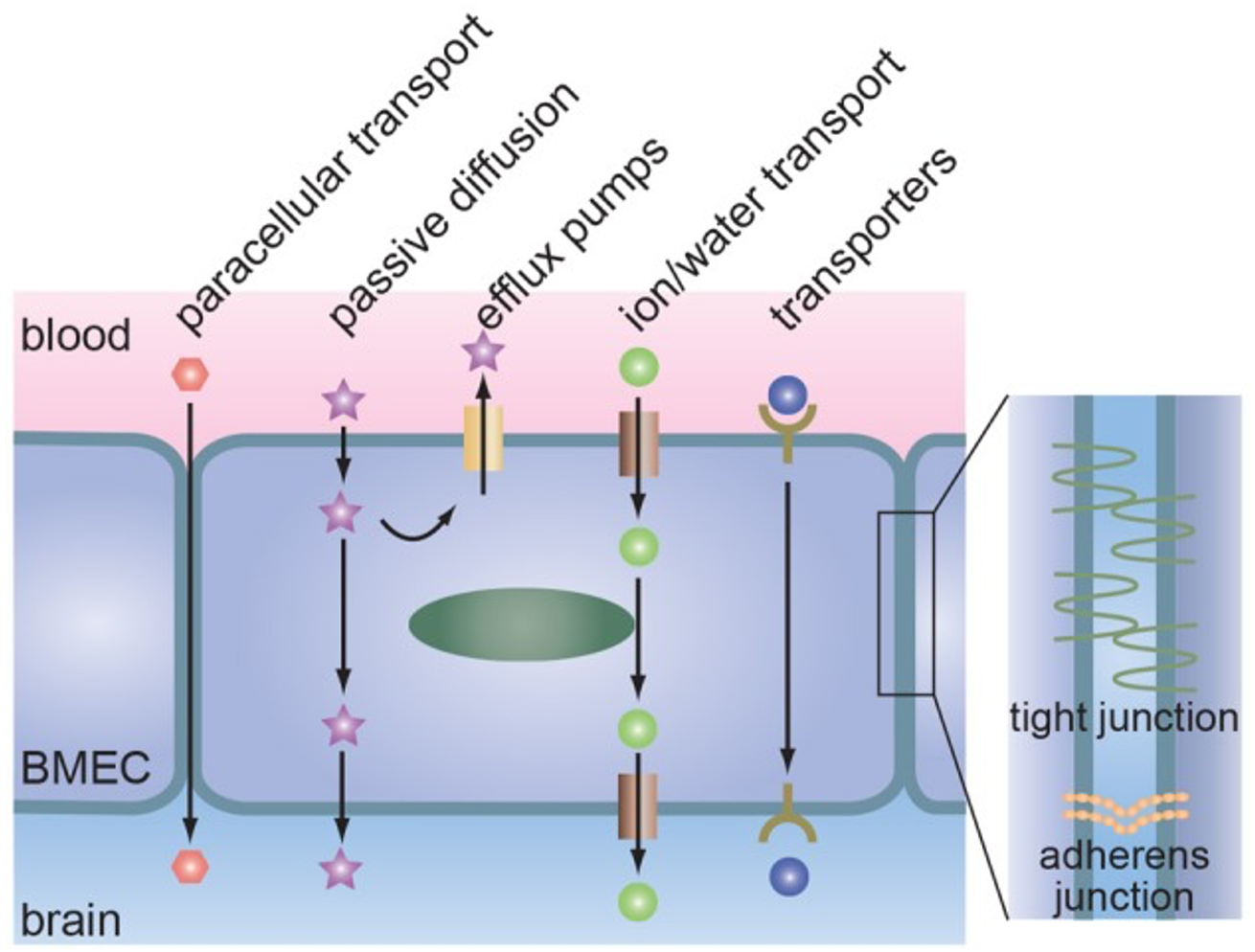
Data
Training Data
Binary labeled training data was acquired through publicly available bioassay data from PubChem. Bioassay records for Amyloid Precursor Protein and Tau were associated with PubChem BioAssays AID1285 and AID1468 [8, 9].
For the APP inhibitory assay 193,714 compounds were tested with only 1,590 (0.82%) showing inhibitory activity. Activity was based on % inhibition of an in-vitro screening assay, where compounds inhibiting expression of APP by 45% or more at 2 uM concentration were labeled as biologically active.
For the Tau fibril inhibitory assay 271,042 compounds were tested, however 19,420 compounds returned inconclusive results and were dropped from the analyses. Out of the remaining 251,982 compounds. 1,025 (0.41%) of these compounds indicated inhibitory activity against the target. Activity was based on a series of dose-response assays and a score using the multiple screenings was calculated. Compounds with scores >= 40 were labeled as biologically active.
There was considerable overlap between the two assays, with 264,965 total compounds tested after combining the datasets and removing duplicates. Thirty-one compounds displayed inhibitory activity against both APP and Tau.
Descriptive Data
ChEMBL and PubChem were used for accessing descriptive data for the compounds in this study, such as molecular weight, the number of aromatic rings, and the solubility of a compound.
Prediction Set
DrugBank (9,468 compounds) includes approved, experimental, investigational, and illicit drugs in their database. 1,027 approved drugs are included in the data set, where the “approved” designation indicates that at some point in time the drug was approved for use somewhere in the world.
Exploratory Data Analysis
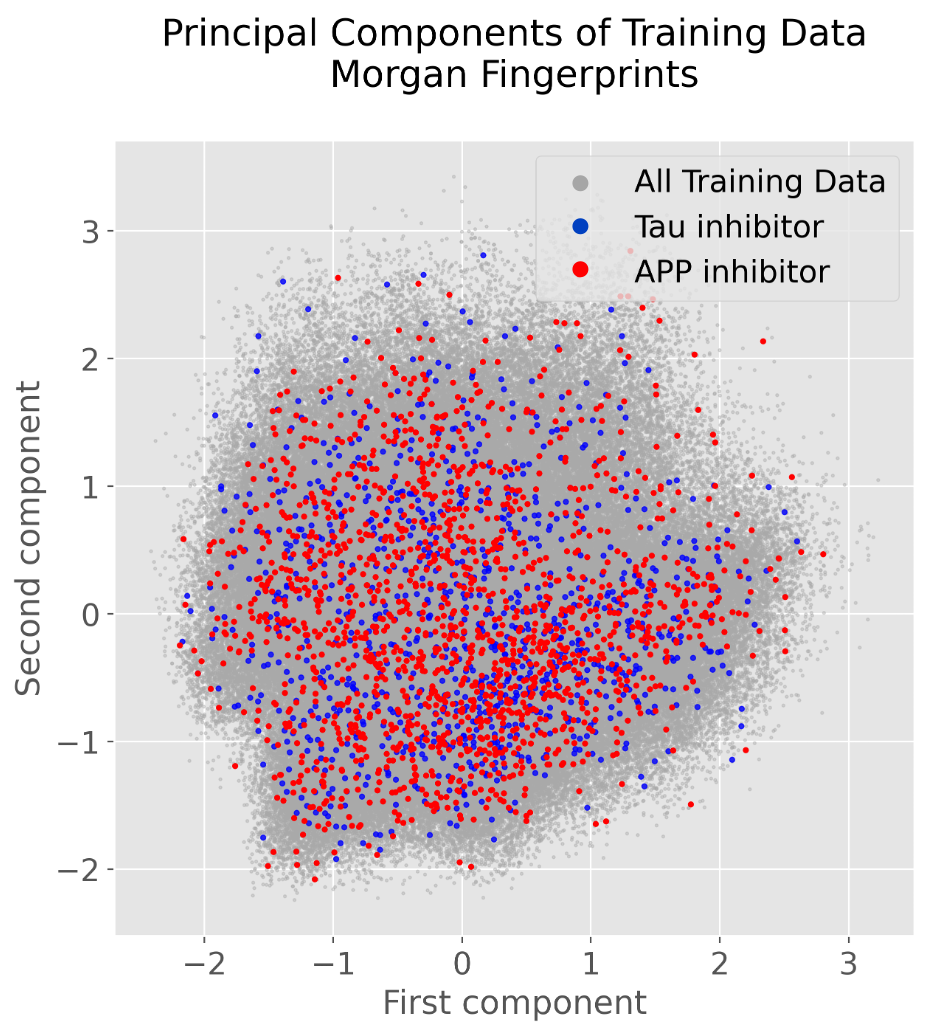
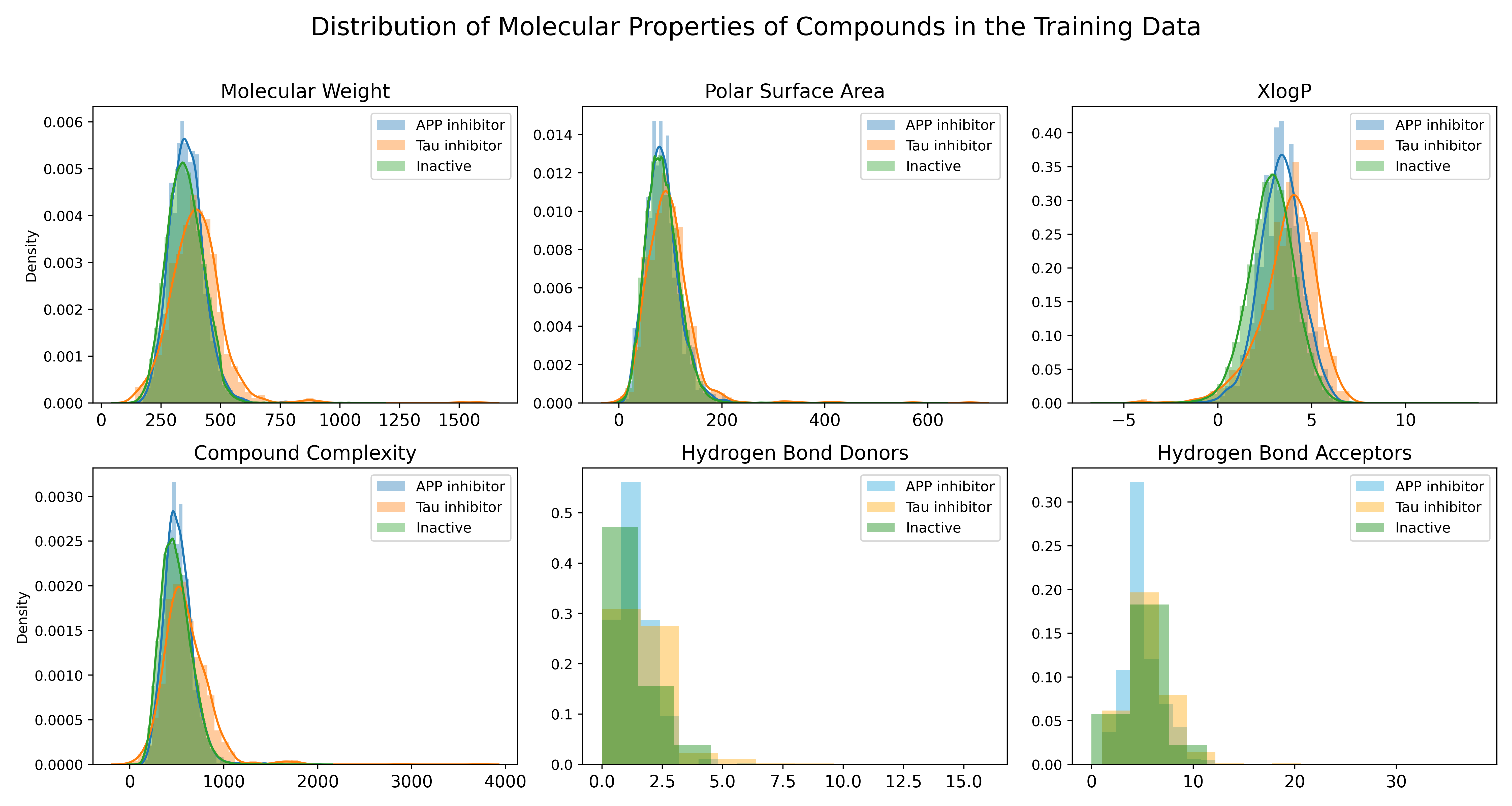
Methods
The project was developed using the Paperspace Cloud Platform to access GPU resources. The models were trained on the NVIDIA RTX 4000 Graphics Card.
The main framework used for development of the analyses was the DeepChem Python library [10]. DeepChem builds on top of PyTorch and Tensorflow deep learning models, making development easier for biological and chemical machine learning. DeepChem provides dozens of deep learning models and architectures that can be used for different classification and regression models.
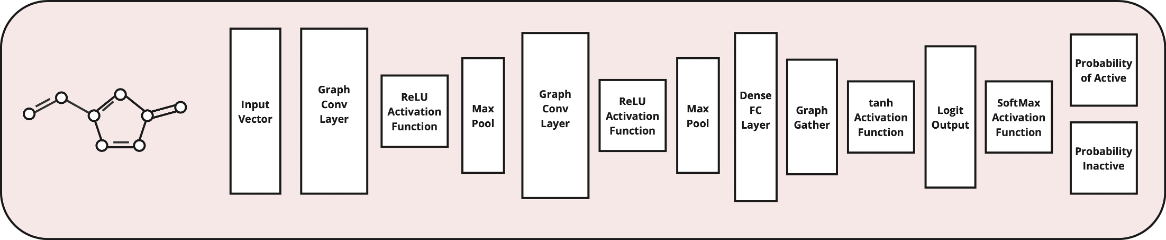
The GraphConvModel, built on top of a Keras Graph Convolution Neural Network Model, was the primary model used for this project. The GraphConvModel has a general architecture like that pictured below, composed of graph convolutional layers, max pooling, and ReLU activation functions. Prior to final probabilities, a dense layer is used to produce outputs based on the trained molecular embedding. The softmax activation function finally converts these outputs into the class probabilities.
 RDKit, one of the most popular cheminformatics Python libraries, was used heavily throughout the project as well.
RDKit, one of the most popular cheminformatics Python libraries, was used heavily throughout the project as well.
Molecular Graph Generation
Molecular graphs are generated using the ConvMolFeaturizer function. SMILES strings are passed into the function and a ConvMol object is returned. This object maintains the adjacency matrix and atom descriptor matrix for the molecule and can be passed directly into the GraphConvModel. The model subsequently extracts the numeric features of the ConvMol object for training and predictions.
Data Preparation
Prior to any training and predictions, compounds were stripped of salts and had their charges neutralized using RDKit.
Subsequently, the PubChem Data was split into 80:10:10 (train: validation: test) sets. The RandomStratifiedSplitter DeepChem function was used to randomly split the assay data while maintaining equivalent active class percentages between the sets. The BalancingTransformer DeepChem function was used to balance the weights of the active and inactive classifications during training. This helps mitigate the consequences of highly class-imbalanced datasets, common in drug-discovery tasks, and very relevant to the data in this project.
Model Architectures
I built on the default parameters of the DeepChem GraphConvModel and expanded my testing from these default parameters. For both protein targets, I performed a random grid search of different model architectures and intermittently measured the precision and recall throughout training. The parameters tested included:
- Batch normalization (True/False)
- Dropout (0, 0.2, 0.5)
- Size of the final fully-connected (dense) layer (128, 256)
-
Size and number of graph convolution layers
- [64, 64]
- [64, 128, 64]
- [64, 64, 64, 64]
- [64, 128, 256, 256, 128, 64]
- [64, 128, 256, 384, 256, 128, 64]
Each target was tested with more than 100 combinations of the parameters for 50-150 epochs. Early stopping was performed if the models did not predict any active classifications after the 10th epoch. Fixed parameters of the models included the use of the Adam optimizer, a learning rate = 0.001, a classification threshold = 0.2 (> 0.2 probability = active classification) and the binary cross-entropy loss function. The softmax activation function was used to calculate the probabilities of each compound being active or inactive against the target protein.
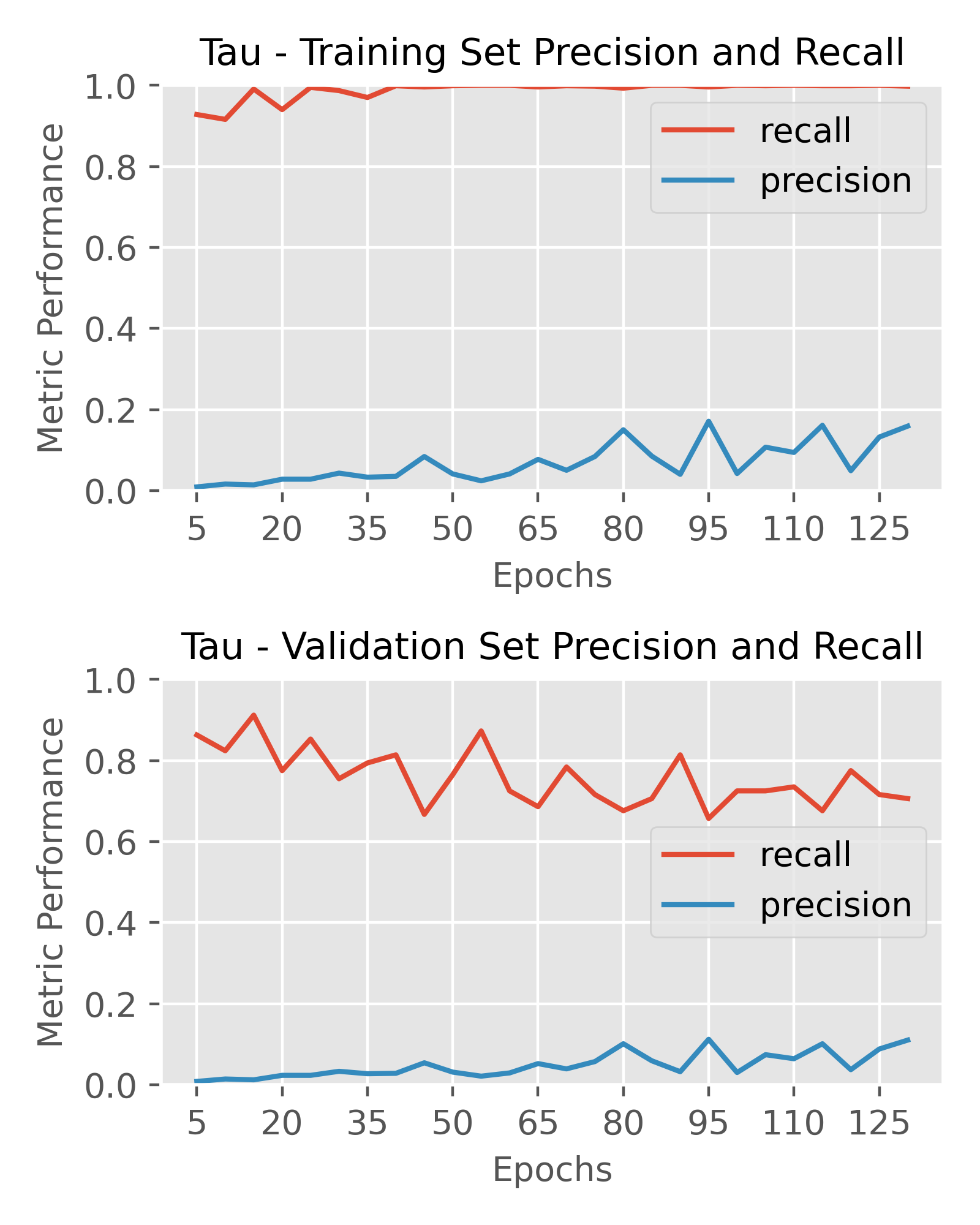
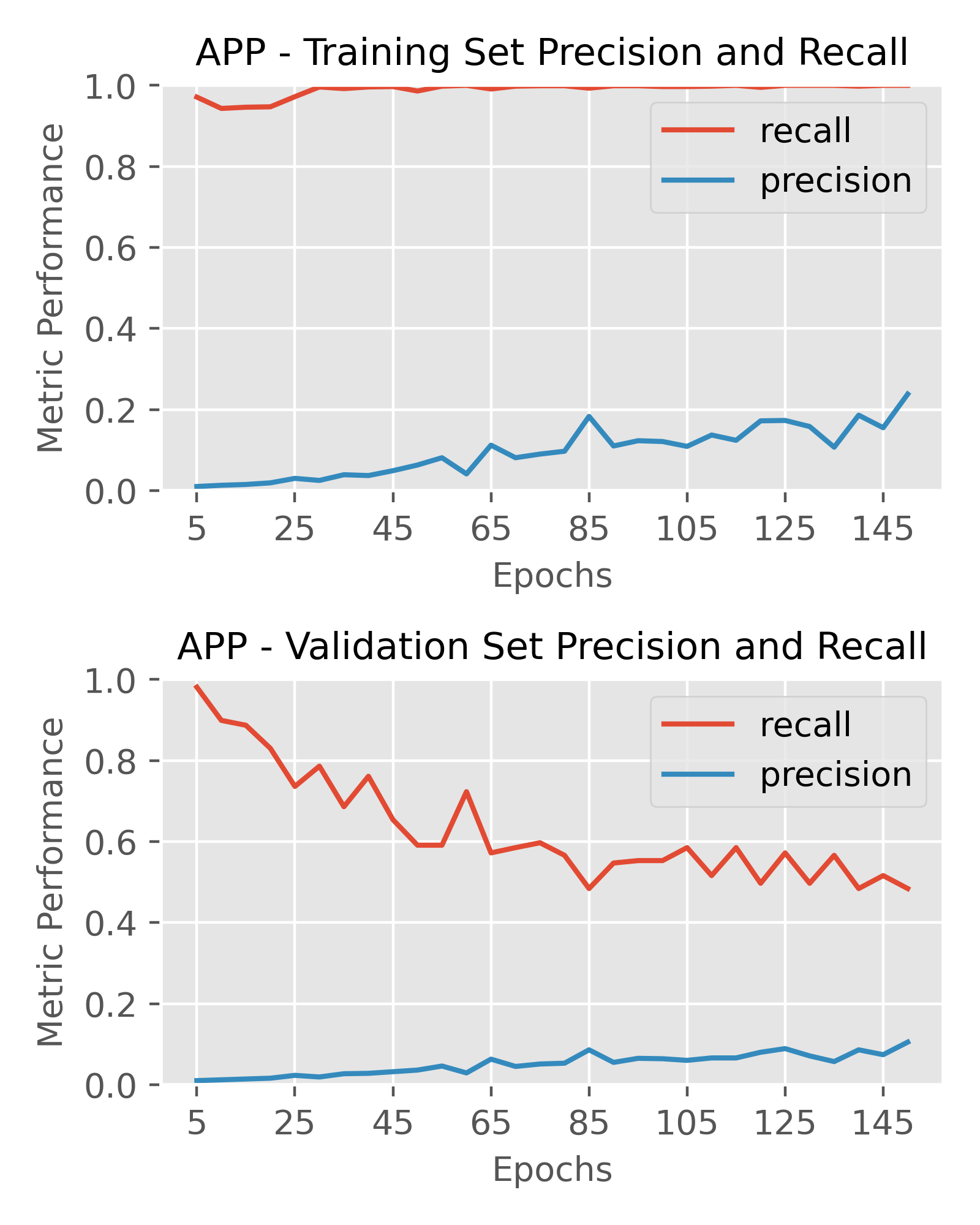
Final Model – Outputs and Performance
After extensive testing of various architectures, I chose the optimal architectures for the targets as follows. A prediction threshold of 0.2 was used for labeling compounds as active or inactive to calculate performance metrics. My primary metric of interest was precision, as I wanted to optimize hit rate and in-vitro screening efficiency, however recall was still important to ensure I wasn’t missing out on too many true positives either.